Plot Repeat Correction Model
plot_repeat_correction_model.RdPlots the results of the repeat correction model for a list of fragments.
Arguments
- fragments_list
A list of fragments class objects obtained from the
call_repeats()function when thecorrection = "repeat"parameter is used.- batch_run_id_subset
A character vector for a subset of batch_sample_id to plot. Or alternatively supply a number to select batch sample by position in alphabetical order.
- n_facet_col
A numeric value indicating the number of columns for faceting in the plot.
Details
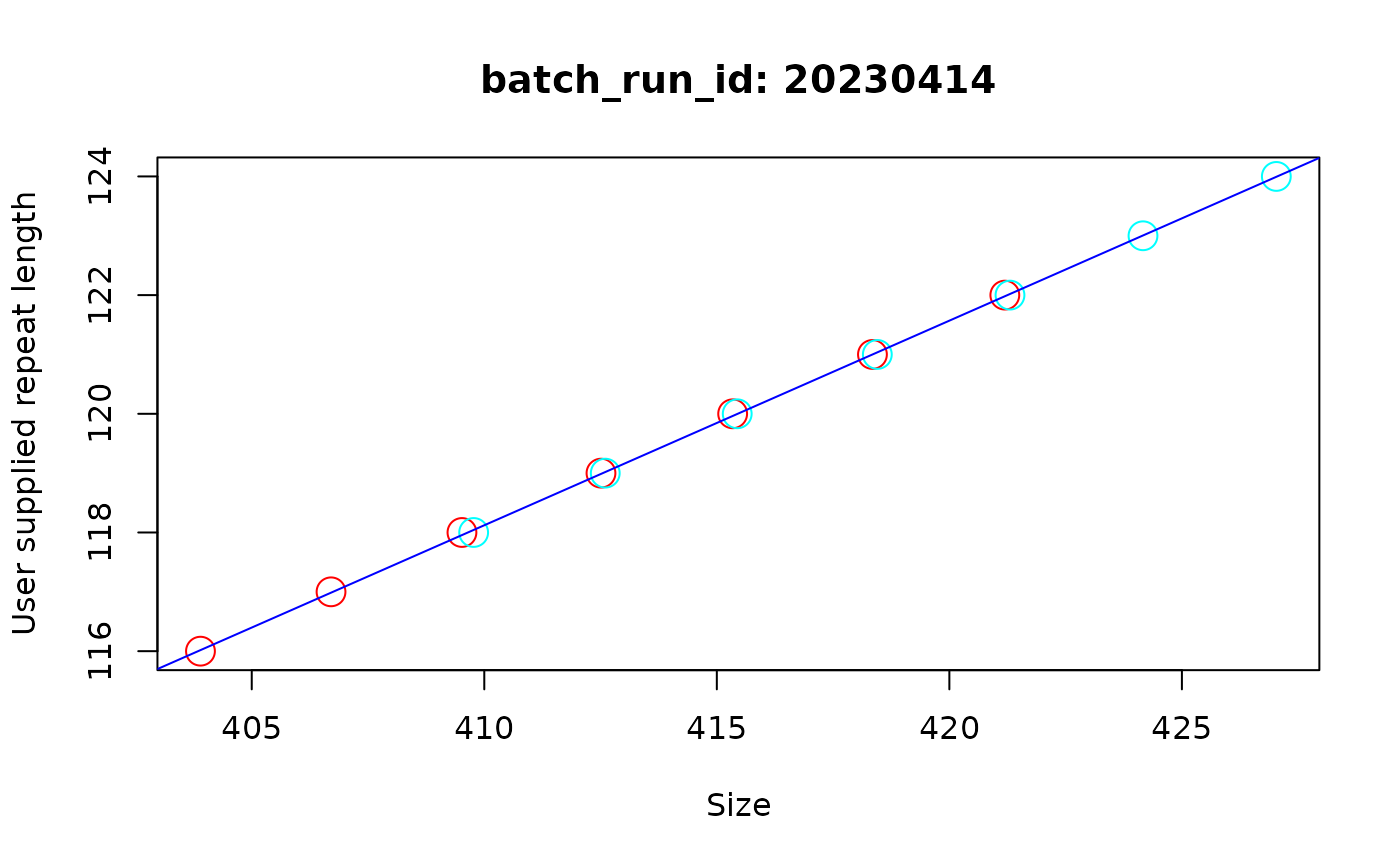
This function makes plots for the model used to correct samples for each batch_run_id. The repeat correction algorithm assigns the user supplied repeat length to the modal peak of the sample, then pulls out a set of robust neighboring peaks to help get enough data to build an accurate linear model for the relationship between base-pair size and repeat length. So on this plot, each dot is an individual peak, with the colour indicating each sample, with the y-axis is the repeat length called from the user-supplied value in the metadata and the value assigned to each peak, with the x-axis showing the corresponding base-pair size.
Examples
fsa_list <- lapply(cell_line_fsa_list, function(x) x$clone())
# import data with read_fsa() to generate an equivalent list to cell_line_fsa_list
fragments_list <- trace(fsa_list, metadata_data.frame = metadata, correction = "repeat")
#> Finding ladders
#>
|
| | 0%
|
|==== | 5%
|
|======= | 11%
|
|=========== | 16%
|
|=============== | 21%
|
|================== | 26%
|
|====================== | 32%
|
|========================== | 37%
|
|============================= | 42%
|
|================================= | 47%
|
|===================================== | 53%
|
|========================================= | 58%
|
|============================================ | 63%
|
|================================================ | 68%
|
|==================================================== | 74%
|
|======================================================= | 79%
|
|=========================================================== | 84%
|
|=============================================================== | 89%
|
|================================================================== | 95%
|
|======================================================================| 100%
#> Finding fragments
#>
|
| | 0%
|
|==== | 5%
|
|======= | 11%
|
|=========== | 16%
|
|=============== | 21%
|
|================== | 26%
|
|====================== | 32%
|
|========================== | 37%
|
|============================= | 42%
|
|================================= | 47%
|
|===================================== | 53%
|
|========================================= | 58%
|
|============================================ | 63%
|
|================================================ | 68%
|
|==================================================== | 74%
|
|======================================================= | 79%
|
|=========================================================== | 84%
|
|=============================================================== | 89%
|
|================================================================== | 95%
|
|======================================================================| 100%
#> Finding alleles
#> Calling repeats
#> Repeat correction model: 4 samples used to build model
#> Repeat correction model: 2.87 bp increase per repeat
#> Assigning index peaks
# traces of bp size shows traces at different sizes
plot_repeat_correction_model(
fragments_list,
batch_run_id_subset = "20230414"
)