Call Repeats for Fragments
call_repeats.RdThis function calls the repeat lengths for a list of fragments.
Arguments
- fragments_list
A list of fragments objects containing fragment data.
- config
A trace_config object generated using
load_config().- ...
additional parameters from any of the functions in the pipeline detailed below may be passed to this function. This overwrites values in the
config. These parameters include:assay_size_without_repeatAn integer specifying the assay size without repeat for repeat calling. This is the length of the sequence flanking the repeat in the PCR product. Default:87.repeat_sizeAn integer specifying the repeat size for repeat calling. Default:3.correctionA character vector of either "batch" to carry out a batch correction from common samples across runs (known repeat length not required), or "repeat" to use samples with validated modal repeat lengths to correct the repeat length. Requires metadata to be added (seeadd_metadata()) with both "batch" and "repeat" requiring"batch_run_id", "batch" requiring ("batch_sample_id") and "repeat" requiring"batch_sample_modal_repeat"(but also benefits from having"batch_sample_id"). Default:"none".force_whole_repeat_unitsA logical value specifying if the peaks should be forced to be whole repeat units apart. Usually the peaks are slightly under the whole repeat unit if left unchanged. Default:FALSE.force_repeat_patternA logical value specifying if the peaks should be re called to fit the specific repeat unit pattern. This requires trace information so you must have started with fsa files. Default:FALSE.force_repeat_pattern_size_periodA numeric value to set the peak periodicity bp size. In fragment analysis, the peaks are usually slightly below the actual repeat unit size, so you can use this value to fine tune what the periodicity should be (eg 3*0.93 = 2.79). Default:2.79.force_repeat_pattern_size_windowA numeric value for the size window when assigning the peak. The algorithm jumps to the predicted scan for the next peak. This value opens a window of the given base pair size neighboring scans to pick the tallest in. Default:0.5.
Details
This function has a lot of different options features for determining the repeat length of your samples. This includes i) an option to force the peaks to be whole repeat units apart, ii) corrections to correct batch effects or accurately call repeat length by comparing to samples of known length, and iii) algorithms or re-calling the peaks to remove any contaminating peaks or shoulder-peaks.
———— correction ————
There are two main correction approaches that are somewhat related: either 'batch' or 'repeat'. Batch correction is relatively simple and just requires you to link samples across batches to correct batch-batch variation in repeat sizes. However, even though the repeat size that is return will be precise, it will not be accurate and underestimates the real repeat length. By contrast, repeat correction can be used to accurately call repeat lengths (which also corrects the batch effects). However, the repeat correction will only be as good as your sample used to call the repeat length so this is a challenging and advanced feature. You need to use a sample that reliably returns the same peak as the modal peak, or you need to be willing to understand the shape of the distribution and manually validate the repeat length of each batch_sample_id for each run.
Batch correction uses common sample(s) across fragment analysis runs to correct systematic batch effects that occur with repeat-containing amplicons in capillary electrophoresis. There are slight fluctuations of size across runs for amplicons containing repeats that result in systematic differences around 1-3 base pairs. So, if samples are to be analyzed for different runs, the absolute bp size is not comparable unless this batch effect is corrected. This is only relevant when the absolute size of a amplicons are compared for grouping metrics as described above (otherwise instability metrics are all relative and it doesn’t matter that there’s systematic batch effects across runs) or when plotting traces from different runs. This correction can be achieved by running a couple of samples in every fragment analysis run, or having a single run that takes a couple of samples from every run together, thereby linking them. These samples are then indicated in the metadata with batch_run_id (to group samples by fragment analysis run) and batch_sample_id (to enable linking samples across batches) (see
add_metadata()). Useplot_batch_correction_samples()to plot the samples before and after correction to make sure that is has worked as expected.Samples with known and validated repeat size can be used to accurately call the repeat length (and therefore also correct batch effects). Similar to batch correction, batch_run_id (to group samples by fragment analysis run) and batch_sample_id (to enable linking samples across batches) are used, but importantly batch_sample_modal_repeat is also set (see
add_metadata()). The batch_sample_modal_repeat is the validated repeat length of the modal repeat of the sample. This validated repeat length is then used to call the repeat length of the modal repeat for each sample (by each batch_run_id). Importantly, this correction requires you to know with confidence the repeat length of the modal peak of the sample. Therefore it's important that the sample used for repeat correction has a clear and prominent modal peak. If the repeat length is very long, it's common for the modal peak of a sample to change so if you use this feature you're going to have to understand the shape of the distribution of your sample and double check that the correct peak has been called as the modal peak after you have usedfind_alleles(). If a different peak is selected as the modal peak than usual, you need to go back to the metadata and adjust the repeat size of the size standard (For example, your size standard sample has been validated to have 120 repeats. You runfind_alleles()and look at the distribution of peaks and notice that the peak one repeat unit higher is the modal peak this time. Therefore, you're going to need to set the batch_sample_modal_repeat as 121 in the metadata just for that batch_run_id. In the other runs you would keep the batch_sample_modal_repeat as 120.). For repeat correction, there are several functions to help visualize and summarize the correction:Use
plot_batch_correction_samples()to visualize the same sample across different batches. This can be helpful to make sure that the correction has worked the same across different runs.Use
plot_repeat_correction_model()to visualize the linear model use to correct repeat length for eachbatch_run_id. This can be helpful to make sure the supplied repeat length of different samples are lining up within each run.Generate a summary table of the predicted repeat length for each sample and the average residuals using
extract_repeat_correction_summary(). This can be helpful to pinpoint the sample(s) that need adjusting.
———— force_whole_repeat_units ————
The force_whole_repeat_units option aims to correct for the systematic underestimation in fragment sizes that occurs in capillary electrophoresis. It is independent to the algorithms described above and can be used in conjunction. It modifies repeat lengths in a way that helps align peaks with the underlying repeat pattern, making the repeat lengths whole units (rather than ~0.9 repeats). The calculated repeat lengths start from the main peak's repeat length and increases in increments of the specified repeat_size in either direction. This option basically enables you to get exactly the same result as expansion_index values calculated from data from Genemapper.
———— force_repeat_pattern ————
This parameter re-calls the peaks based on specified (force_repeat_pattern_size_period) periodicity of the peaks. The main application of this algorithm is to solve the issue of contaminating peaks in the expected regular pattern of peaks. We can use the periodicity to jump between peaks and crack open a window (force_repeat_pattern_size_window) to then pick out the tallest scan in the window.
Examples
fsa_list <- lapply(cell_line_fsa_list[c(16:19)], function(x) x$clone())
config <- load_config()
trace:::find_ladders(fsa_list, config, show_progress_bar = FALSE)
trace:::find_fragments(
fsa_list,
config,
min_bp_size = 300,
show_progress_bar = FALSE
)
trace:::find_alleles(fsa_list, config)
trace:::add_metadata(fsa_list,
metadata[c(16:19), ]
)
# Simple conversion from bp size to repeat size
trace:::call_repeats(
fsa_list,
config,
assay_size_without_repeat = 87,
repeat_size = 3
)
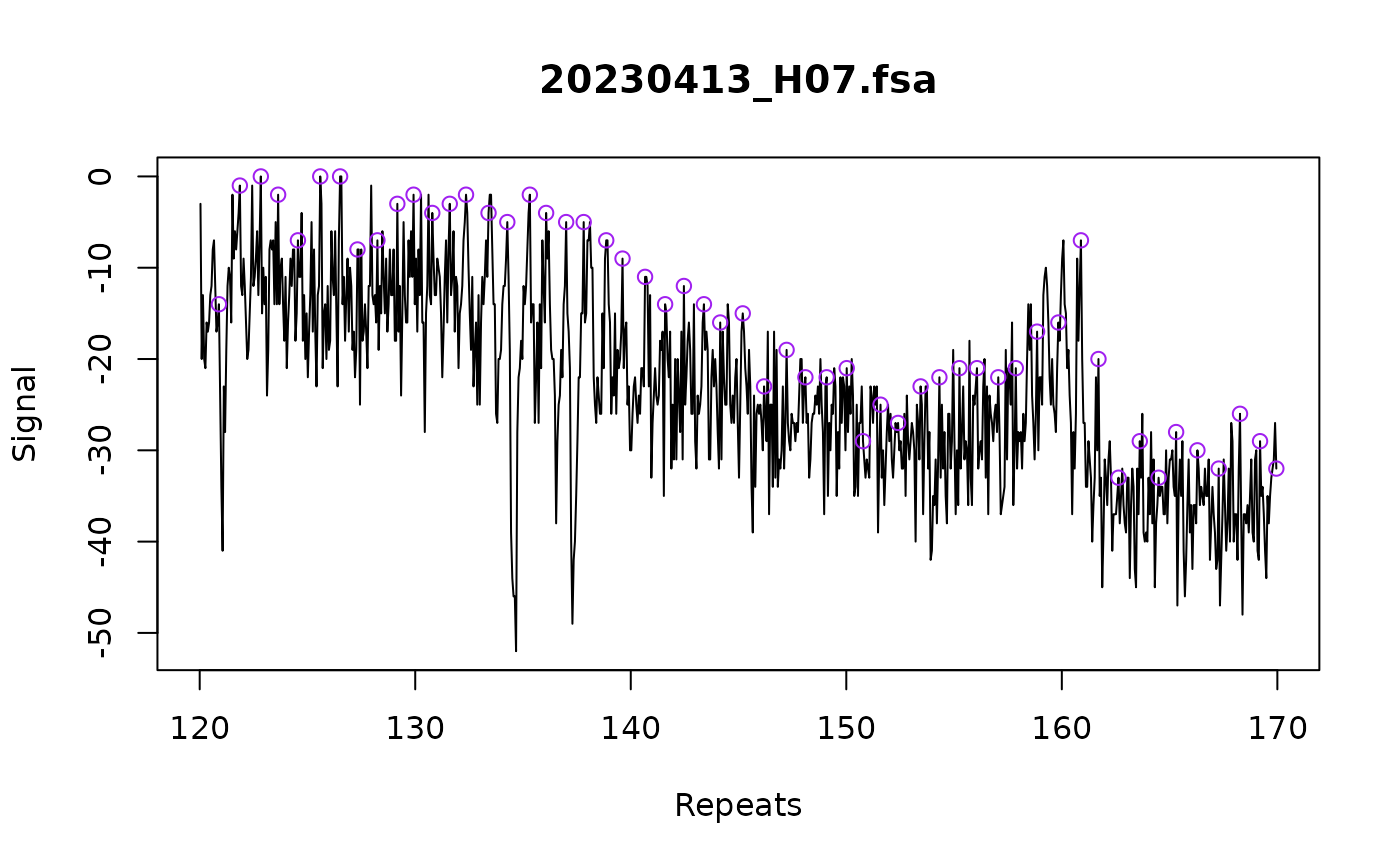
plot_traces(fsa_list[1], xlim = c(120, 170))
#> Warning: no non-missing arguments to max; returning -Inf
 # Use force_whole_repeat_units algorithm to make sure called
# repeats are the exact number of bp apart
trace:::call_repeats(
fsa_list,
config,
force_whole_repeat_units = TRUE,
assay_size_without_repeat = 87,
repeat_size = 3
)
plot_traces(fsa_list[1], xlim = c(120, 170))
#> Warning: no non-missing arguments to max; returning -Inf
# Use force_whole_repeat_units algorithm to make sure called
# repeats are the exact number of bp apart
trace:::call_repeats(
fsa_list,
config,
force_whole_repeat_units = TRUE,
assay_size_without_repeat = 87,
repeat_size = 3
)
plot_traces(fsa_list[1], xlim = c(120, 170))
#> Warning: no non-missing arguments to max; returning -Inf
 # apply batch correction
trace:::call_repeats(
fsa_list,
config,
correction = "batch",
assay_size_without_repeat = 87,
repeat_size = 3
)
#> Correcting batch effects
plot_traces(fsa_list[1], xlim = c(120, 170))
#> Warning: no non-missing arguments to max; returning -Inf
# apply batch correction
trace:::call_repeats(
fsa_list,
config,
correction = "batch",
assay_size_without_repeat = 87,
repeat_size = 3
)
#> Correcting batch effects
plot_traces(fsa_list[1], xlim = c(120, 170))
#> Warning: no non-missing arguments to max; returning -Inf
 # apply repeat correction
trace:::call_repeats(
fsa_list,
config,
correction = "repeat",
assay_size_without_repeat = 87,
repeat_size = 3
)
#> Repeat correction model: 4 samples used to build model
#> Repeat correction model: 2.87 bp increase per repeat
plot_traces(fsa_list[1], xlim = c(120, 170))
# apply repeat correction
trace:::call_repeats(
fsa_list,
config,
correction = "repeat",
assay_size_without_repeat = 87,
repeat_size = 3
)
#> Repeat correction model: 4 samples used to build model
#> Repeat correction model: 2.87 bp increase per repeat
plot_traces(fsa_list[1], xlim = c(120, 170))
 #ensure only periodic peaks are called
trace:::call_repeats(
fsa_list,
config,
force_repeat_pattern_size_period = 2.75,
assay_size_without_repeat = 87,
repeat_size = 3
)
plot_traces(fsa_list[1], xlim = c(120, 170))
#> Warning: no non-missing arguments to max; returning -Inf
#ensure only periodic peaks are called
trace:::call_repeats(
fsa_list,
config,
force_repeat_pattern_size_period = 2.75,
assay_size_without_repeat = 87,
repeat_size = 3
)
plot_traces(fsa_list[1], xlim = c(120, 170))
#> Warning: no non-missing arguments to max; returning -Inf